InVivoMAb anti-mouse CD4
Product Details
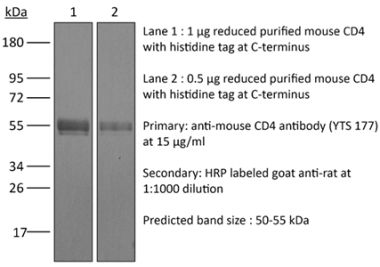
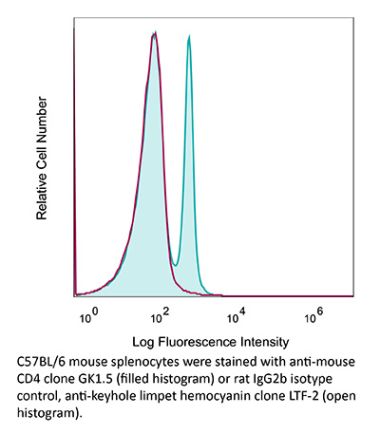
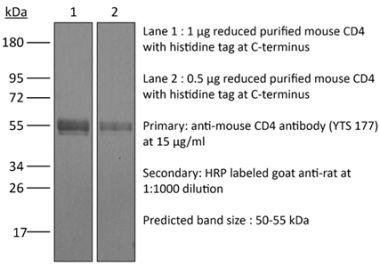
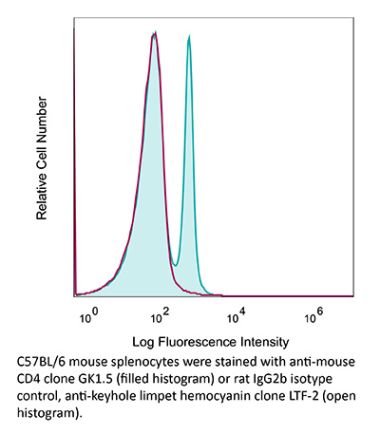
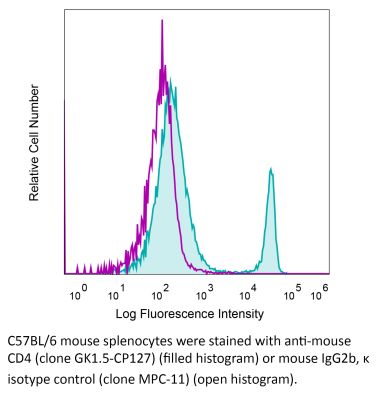
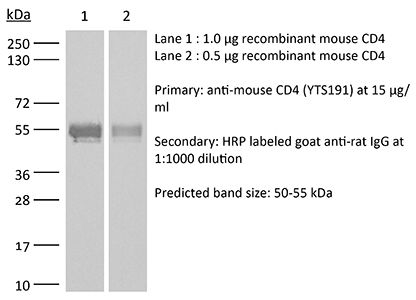
The YTS 191 monoclonal antibody reacts with mouse CD4. The CD4 antigen is a 55 kDa type I cell-surface membrane glycoprotein belonging to the immunoglobulin superfamily. CD4 acts as a co-receptor which in cooperation with the T cell receptor (TCR) interacts with class II MHC molecules displayed by antigen presenting cells (APC). CD4 is expressed by the majority of thymocytes, most helper T cells, a subset of NK-T cells and weakly by dendritic cells and macrophages. CD4 plays an important role in the development of T cells and is required for mature T cells to function optimally. The YTS 191 antibody has been shown to compete with clones GK1.5 and YTS 177 for CD4 binding.Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Not available or unknown |
| Reported Applications | in vivo CD4+ T cell depletion |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10950382 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo CD4+ T cell depletion
Kreiter, S., et al. (2015). "Mutant MHC class II epitopes drive therapeutic immune responses to cancer" Nature 520(7549): 692-696. PubMed
Tumour-specific mutations are ideal targets for cancer immunotherapy as they lack expression in healthy tissues and can potentially be recognized as neo-antigens by the mature T-cell repertoire. Their systematic targeting by vaccine approaches, however, has been hampered by the fact that every patient’s tumour possesses a unique set of mutations (‘the mutanome’) that must first be identified. Recently, we proposed a personalized immunotherapy approach to target the full spectrum of a patient’s individual tumour-specific mutations. Here we show in three independent murine tumour models that a considerable fraction of non-synonymous cancer mutations is immunogenic and that, unexpectedly, the majority of the immunogenic mutanome is recognized by CD4(+) T cells. Vaccination with such CD4(+) immunogenic mutations confers strong antitumour activity. Encouraged by these findings, we established a process by which mutations identified by exome sequencing could be selected as vaccine targets solely through bioinformatic prioritization on the basis of their expression levels and major histocompatibility complex (MHC) class II-binding capacity for rapid production as synthetic poly-neo-epitope messenger RNA vaccines. We show that vaccination with such polytope mRNA vaccines induces potent tumour control and complete rejection of established aggressively growing tumours in mice. Moreover, we demonstrate that CD4(+) T cell neo-epitope vaccination reshapes the tumour microenvironment and induces cytotoxic T lymphocyte responses against an independent immunodominant antigen in mice, indicating orchestration of antigen spread. Finally, we demonstrate an abundance of mutations predicted to bind to MHC class II in human cancers as well by employing the same predictive algorithm on corresponding human cancer types. Thus, the tailored immunotherapy approach introduced here may be regarded as a universally applicable blueprint for comprehensive exploitation of the substantial neo-epitope target repertoire of cancers, enabling the effective targeting of every patient’s tumour with vaccines produced ‘just in time’.
in vivo CD4+ T cell depletion
Burrack, K. S., et al. (2015). "Myeloid Cell Arg1 Inhibits Control of Arthritogenic Alphavirus Infection by Suppressing Antiviral T Cells" PLoS Pathog 11(10): e1005191. PubMed
Arthritogenic alphaviruses, including Ross River virus (RRV) and chikungunya virus (CHIKV), are responsible for explosive epidemics involving millions of cases. These mosquito-transmitted viruses cause inflammation and injury in skeletal muscle and joint tissues that results in debilitating pain. We previously showed that arginase 1 (Arg1) was highly expressed in myeloid cells in the infected and inflamed musculoskeletal tissues of RRV- and CHIKV-infected mice, and specific deletion of Arg1 from myeloid cells resulted in enhanced viral control. Here, we show that Arg1, along with other genes associated with suppressive myeloid cells, is induced in PBMCs isolated from CHIKV-infected patients during the acute phase as well as the chronic phase, and that high Arg1 expression levels were associated with high viral loads and disease severity. Depletion of both CD4 and CD8 T cells from RRV-infected Arg1-deficient mice restored viral loads to levels detected in T cell-depleted wild-type mice. Moreover, Arg1-expressing myeloid cells inhibited virus-specific T cells in the inflamed and infected musculoskeletal tissues, but not lymphoid tissues, following RRV infection in mice, including suppression of interferon-gamma and CD69 expression. Collectively, these data enhance our understanding of the immune response following arthritogenic alphavirus infection and suggest that immunosuppressive myeloid cells may contribute to the duration or severity of these debilitating infections.
in vivo CD4+ T cell depletion
Wensveen, F. M., et al. (2015). "NK cells link obesity-induced adipose stress to inflammation and insulin resistance" Nat Immunol 16(4): 376-385. PubMed
An important cause of obesity-induced insulin resistance is chronic systemic inflammation originating in visceral adipose tissue (VAT). VAT inflammation is associated with the accumulation of proinflammatory macrophages in adipose tissue, but the immunological signals that trigger their accumulation remain unknown. We found that a phenotypically distinct population of tissue-resident natural killer (NK) cells represented a crucial link between obesity-induced adipose stress and VAT inflammation. Obesity drove the upregulation of ligands of the NK cell-activating receptor NCR1 on adipocytes; this stimulated NK cell proliferation and interferon-gamma (IFN-gamma) production, which in turn triggered the differentiation of proinflammatory macrophages and promoted insulin resistance. Deficiency of NK cells, NCR1 or IFN-gamma prevented the accumulation of proinflammatory macrophages in VAT and greatly ameliorated insulin sensitivity. Thus NK cells are key regulators of macrophage polarization and insulin resistance in response to obesity-induced adipocyte stress.
in vivo CD4+ T cell depletion
Yamada, D. H., et al. (2015). "Suppression of Fcgamma-receptor-mediated antibody effector function during persistent viral infection" Immunity 42(2): 379-390. PubMed
Understanding how viruses subvert host immunity and persist is essential for developing strategies to eliminate infection. T cell exhaustion during chronic viral infection is well described, but effects on antibody-mediated effector activity are unclear. Herein, we show that increased amounts of immune complexes generated in mice persistently infected with lymphocytic choriomeningitis virus (LCMV) suppressed multiple Fcgamma-receptor (FcgammaR) functions. The high amounts of immune complexes suppressed antibody-mediated cell depletion, therapeutic antibody-killing of LCMV infected cells and human CD20-expressing tumors, as well as reduced immune complex-mediated cross-presentation to T cells. Suppression of FcgammaR activity was not due to inhibitory FcgammaRs or high concentrations of free antibody, and proper FcgammaR functions were restored when persistently infected mice specifically lacked immune complexes. Thus, we identify a mechanism of immunosuppression during viral persistence with implications for understanding effective antibody activity aimed at pathogen control.
in vivo CD4+ T cell depletion
Steel, C. D., et al. (2014). "Role of peripheral immune response in microglia activation and regulation of brain chemokine and proinflammatory cytokine responses induced during VSV encephalitis" J Neuroimmunol 267(1-2): 50-60. PubMed
We report herein that neuroinvasion by vesicular stomatitis virus (VSV) activates microglia and induces a peripheral dendritic cell (DC)-dependent inflammatory response in the central nervous system (CNS). VSV neuroinvasion rapidly induces multiple brain chemokine and proinflammatory cytokine mRNAs that display bimodal kinetics. Peripheral DC ablation or T cell depletion suppresses the second wave of this response demonstrating that infiltrating T cells are primarily responsible for the bimodal characteristics of this response. The robust infiltrate associated with VSV encephalitis likely depends on sustained production of brain CCL19 and CCR7 expression on infiltrating inflammatory cells.
in vivo CD4+ T cell depletion
Kish, D. D., et al. (2011). "Hapten application to the skin induces an inflammatory program directing hapten-primed effector CD8 T cell interaction with hapten-presenting endothelial cells" J Immunol 186(4): 2117-2126. PubMed
Contact hypersensitivity is a CD8 T cell-mediated response to hapten sensitization and challenge of the skin. Effector CD8 T cell recruitment into the skin parenchyma to elicit the response to hapten challenge requires prior CXCL1/KC-directed neutrophil infiltration within 3-6 h after challenge and is dependent on IFN-gamma and IL-17 produced by the hapten-primed CD8 T cells. Mechanisms directing hapten-primed CD8 T cell localization and activation in the Ag challenge site to induce this early CXCL1 production in response to 2,4-dinitrofluorobenzene were investigated. Both TNF-alpha and IL-17, but not IFN-gamma, mRNA was detectable within 1 h of hapten challenge of sensitized mice and increased thereafter. Expression of ICAM-1 was observed by 1 h after challenge of sensitized and nonsensitized mice and was dependent on TNF-alpha. The induction of IL-17, IFN-gamma, and CXCL1 in the challenge site was not observed when ICAM-1 was absent or neutralized by specific Ab. During the elicitation of the contact hypersensitivity response, endothelial cells expressed ICAM-1 and produced CXCL1 suggesting this as the site of CD8 T cell localization and activation. Endothelial cells isolated from challenged skin of naive and sensitized mice had acquired the hapten and the ability to activate hapten-primed CD8 T cell cytokine production. These results indicate that hapten application to the skin of sensitized animals initiates an inflammatory response promoting hapten-primed CD8 T cell localization to the challenge site through TNF-alpha-induced ICAM-1 expression and CD8 T cell activation to produce IFN-gamma and IL-17 through endothelial cell presentation of hapten.
in vivo CD4+ T cell depletion
Fahey, L. M., et al. (2011). "Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells" J Exp Med 208(5): 987-999. PubMed
CD4 T cell responses are crucial to prevent and control viral infection; however, virus-specific CD4 T cell activity is considered to be rapidly lost during many persistent viral infections. This is largely caused by the fact that during viral persistence CD4 T cells do not produce the classical Th1 cytokines associated with control of acute viral infections. Considering that CD4 T cell help is critical for both CD8 T cell and B cell functions, it is unclear how CD4 T cells can lose responsiveness but continue to sustain long-term control of persistent viral replication. We now demonstrate that CD4 T cell function is not extinguished as a result of viral persistence. Instead, viral persistence and prolonged T cell receptor stimulation progressively redirects CD4 T cell development away from the Th1 response induced during an acute infection toward T follicular helper cells. Importantly, this sustained CD4 T cell functionality is critical to maintain immunity and ultimately aid in the control of persistent viral infection.
in vivo CD4+ T cell depletion
Kish, D. D., et al. (2009). "CD8 T cells producing IL-17 and IFN-gamma initiate the innate immune response required for responses to antigen skin challenge" J Immunol 182(10): 5949-5959. PubMed
Effector CD8 T cell recruitment into the skin in response to Ag challenge requires prior CXCL1/KC-directed neutrophil infiltration. Mechanisms inducing CXCL1 production and the dynamics of neutrophil-CD8 T cell interactions during elicitation of Ag-specific responses in the skin were investigated. CXCL1 and CXCL2/MIP-2 were produced within 3-6 h of Ag challenge at 10-fold higher levels in skin challenge sites of Ag-sensitized vs nonsensitized mice. In the challenge sites of sensitized mice this production decreased at 6-9 h postchallenge to near the levels observed in skin challenge sites of nonsensitized mice but rose to a second peak 12 h after challenge. The elevated early neutrophil chemoattractant production at 3-6 h after skin challenge of sensitized animals required both IFN-gamma and IL-17, produced by distinct populations of Ag-primed CD8 T cells in response to Ag challenge. Although induced by the Ag-primed CD8 T cells, the early CXCL1 and CXCL2 production was accompanied by neutrophil but not CD8 T cell infiltration into the skin Ag challenge site. Infiltration of the CD8 T cells into the challenge site was not observed until 18-24 h after challenge. These results demonstrate an intricate series of early interactions between Ag-specific and innate immune components that regulate the sequential infiltration of neutrophils and then effector T cells into the skin to mediate an immune response.
- Cancer Research,
- Immunology and Microbiology
Combination therapy with alisertib enhances the anti-tumor immunity induced by a liver cancer vaccine.
In IScience on 18 April 2025 by Xue, F., Liu, J., et al.
Alisertib is a potent aurora A kinase inhibitor in clinical trials for cancer treatment, but its efficacy on cancer vaccines remains unclear. Here, we developed a DNA vaccine targeting glypican-3 (pGPC3) and evaluated its efficacy with alisertib in hepatocellular carcinoma (HCC) models. The combination therapy of pGPC3 vaccine and alisertib significantly inhibited subcutaneous tumor growth, enhanced the induction and maturation of CD11c+ and CD8+CD11c+ dendritic cells (DCs), and expanded tumor-specific CD8+ T cell responses. CD8+ T cell depletion abolished the anti-tumor effects, underscoring the essential role of functional CD8+ T cell responses. Moreover, the combined treatment promoted memory CD8+ T cell induction, providing long-term protection. In liver orthotopic tumor models, the combination of pGPC3 vaccine and alisertib demonstrated potent therapeutic efficacy through CD8+ T cell responses. These results indicate that alisertib enhances the pGPC3 vaccine's therapeutic effect, offering a promising strategy for HCC treatment. © 2025 The Author(s).
- Cancer Research,
- Immunology and Microbiology
ULBP2 Promotes Tumor Progression by Suppressing NKG2D-Mediated Anti-Tumor Immunity.
In International Journal of Molecular Sciences on 24 March 2025 by Yamane, K., Yamaguchi, K., et al.
UL-16 binding protein 2 (ULBP2), a human NKG2D ligand, has been identified as a poor prognostic factor in several cancers based on recent comprehensive analyses of immune-related genes using the Cancer Genome Atlas datasets. Despite its clinical significance, the functional role of ULBP2 in vivo remains largely unknown. In this study, we investigated the role of ULBP2 in modulating anti-tumor immunity using murine melanoma cell lines engineered to stably express surface-expressed or soluble ULBP2. Subcutaneous transplantation of ULBP2-expressing melanoma cells into syngeneic mice resulted in accelerated tumor growth, mediated by surface-expressed ULBP2, through the suppression of NKG2D-dependent immune responses. In vitro experiments revealed that sustained exposure to tumor-expressed ULBP2 reduced NKG2D expression and cytotoxic activity of splenocytes. In contrast, soluble ULBP2 did not significantly affect tumor growth or immune responses. These findings suggest that surface-expressed ULBP2 plays a pivotal role in tumor immune evasion and highlight its potential as a therapeutic target to enhance anti-tumor immunity.
- Mus musculus (House mouse),
- Cancer Research,
- Genetics,
- Immunology and Microbiology
Lipid nanoparticles deliver DNA-encoded biologics and induce potent protective immunity.
In Molecular Cancer on 13 January 2025 by Chai, D., Wang, J., et al.
Lipid nanoparticles (LNPs) for mRNA delivery have advanced significantly, but LNP-mediated DNA delivery still faces clinical challenges. This study compared various LNP formulations for delivering DNA-encoded biologics, assessing their expression efficacy and the protective immunity generated by LNP-encapsulated DNA in different models. The LNP formulation used in Moderna's Spikevax mRNA vaccine (LNP-M) demonstrated a stable nanoparticle structure, high expression efficiency, and low toxicity. Notably, a DNA vaccine encoding the spike protein, delivered via LNP-M, induced stronger antigen-specific antibody and T cell immune responses compared to electroporation. Single-cell RNA sequencing (scRNA-seq) analysis revealed that the LNP-M/pSpike vaccine enhanced CD80 activation signaling in CD8+ T cells, NK cells, macrophages, and DCs, while reducing the immunosuppressive signals. The enrichment of TCR and BCR by LNP-M/pSpike suggested an increase in immune response specificity and diversity. Additionally, LNP-M effectively delivered DNA-encoded antigens, such as mouse PD-L1 and p53R172H, or monoclonal antibodies targeting mouse PD1 and human p53R282W. This approach inhibited tumor growth or metastasis in several mouse models. The long-term anti-tumor effects of LNP-M-delivered anti-p53R282W antibody relied on memory CD8+ T cell responses and enhanced MHC-I signaling from APCs to CD8+ T cells. These results highlight LNP-M as a promising and effective platform for delivering DNA-based vaccines and cancer immunotherapies. © 2025. The Author(s).
- Cancer Research,
- Immunology and Microbiology
Developing an Effective Therapeutic HPV Vaccine to Eradicate Large Tumors by Genetically Fusing Xcl1 and Incorporating IL-9 as Molecular Adjuvants.
In Vaccines on 9 January 2025 by Sun, Z., Wu, Z., et al.
Human papillomavirus (HPV) is a prevalent infection affecting both men and women, leading to various cytological lesions. Therapeutic vaccines mount a HPV-specific CD8+ cytotoxic T lymphocyte response, thus clearing HPV-infected cells. However, no therapeutic vaccines targeting HPV are currently approved for clinical treatment due to limited efficacy. Our goal is to develop a vaccine that can effectively eliminate tumors caused by HPV. We genetically fused the chemokine XCL1 with the E6 and E7 proteins of HPV16 to target cDC1 and enhance the vaccine-induced cytotoxic T cell response, ultimately developing a DNA vaccine. Additionally, we screened various interleukins and identified IL-9 as an effective molecular adjuvant for our DNA vaccine. The fusion of Xcl1 significantly improved the quantity and quality of the specific CD8+ T cells. The fusion of Xcl1 also increased immune cell infiltration into the tumor microenvironment. The inclusion of IL-9 significantly elevated the vaccine-induced specific T cell response and enhanced anti-tumor efficacy. IL-9 promotes the formation of central memory T cells. the fusion of Xcl1 and the use of IL-9 as a molecular adjuvant represent promising strategies for vaccine development.
- In Vivo,
- Mus musculus (House mouse),
- Biochemistry and Molecular biology,
- Cancer Research,
- Cell Biology,
- Immunology and Microbiology
Napabucasin-loaded PLGA nanoparticles trigger anti-HCC immune responses by metabolic reprogramming of tumor-associated macrophages.
In Journal of Translational Medicine on 20 December 2024 by Song, Z., Chen, H., et al.
JAK/STAT3 is one of the critical signaling pathways involved in the occurrence and development of hepatocellular carcinoma (HCC). BBI608 (Napabucasin), as a novel small molecule inhibitor of STAT3, has shown previously excellent anti-HCC effects in vitro and in mouse models. However, low bioavailability, high cytotoxicity and other shortcomings limit its clinical application. In this study, PLGA was selected to prepare Napabucasin PLGA nanoparticles (NPs) by solvent evaporation method, overcoming these limitations and improving the passive targeting effect that nanoparticle mediated. Base on this, we systematically evaluated the anti-HCC effect of Napabucasin-PLGA NPs and explored the underlying mechanisms. Napabucasin-PLGA NPs were prepared by solvent evaporation method. CCK-8 assay, Annexin V/PI double staining, RT-qPCR, colony formation assay, and Western blotting were performed to evaluate the anti-HCC effect of Napabucasin-PLGA NPs in vitro. Proliferation assay and migration assay were used to detect the effects of Napabucasin-PLGA NPs-treated HCC-TAMs on tumor biological characteristics of HCC cells. Flow cytometry was used to detect anti-HCC immune responses induced by Napabucasin-PLGA NPs in vivo. Our results demonstrated that Napabucasin-PLGA NPs could improve the bioavailability of Napabucasin and enhance Napabucasin-mediated the anti-HCC effects in vitro and in vivo with no significant drug toxicity. In addition to the direct inhibitory effects on the tumor biological characteristics of HCC cells, Napabucasin-PLGA NPs could promote the polarization of macrophages from tumor-promoting M2-type to anti-tumor M1-type, improving the tumor immune microenvironment and augmenting T cell-mediated anti-tumor responses. The underlining mechanisms showed Napabucasin-PLGA NPs suppressed the STAT3/FAO signaling axis in HCC-induced tumor-associated macrophages (TAMs). These findings demonstrated Napabucasin-PLGA NPs is a potential therapeutic candidate for HCC, and provided a new theoretical and experimental basis for further development and clinical application of Napabucasin. © 2024. The Author(s).
- Mus musculus (House mouse),
- Immunology and Microbiology
Picrasidine S Induces cGAS-Mediated Cellular Immune Response as a Novel Vaccine Adjuvant.
In Advanced Science (Weinheim, Baden-Wurttemberg, Germany) on 1 August 2024 by Ding, X., Sun, M., et al.
New adjuvants that trigger cellular immune responses are urgently needed for the effective development of cancer and virus vaccines. Motivated by recent discoveries that show activation of type I interferon (IFN-I) signaling boosts T cell immunity, this study proposes that targeting this pathway can be a strategic approach to identify novel vaccine adjuvants. Consequently, a comprehensive chemical screening of 6,800 small molecules is performed, which results in the discovery of the natural compound picrasidine S (PS) as an IFN-I inducer. Further analysis reveals that PS acts as a powerful adjuvant, significantly enhancing both humoral and cellular immune responses. At the molecular level, PS initiates the activation of the cGAS-IFN-I pathway, leading to an enhanced T cell response. PS vaccination notably increases the population of CD8+ central memory (TCM)-like cells and boosts the CD8+ T cell-mediated anti-tumor immune response. Thus, this study identifies PS as a promising candidate for developing vaccine adjuvants in cancer prevention. © 2024 The Author(s). Advanced Science published by Wiley‐VCH GmbH.
- Mus musculus (House mouse),
- Immunology and Microbiology,
- Neuroscience,
- Pathology
T cell-mediated microglial activation triggers myelin pathology in a mouse model of amyloidosis.
In Nature Neuroscience on 1 August 2024 by Kedia, S., Ji, H., et al.
Age-related myelin damage induces inflammatory responses, yet its involvement in Alzheimer's disease remains uncertain, despite age being a major risk factor. Using a mouse model of Alzheimer's disease, we found that amyloidosis itself triggers age-related oligodendrocyte and myelin damage. Mechanistically, CD8+ T cells promote the progressive accumulation of abnormally interferon-activated microglia that display myelin-damaging activity. Thus, our data suggest that immune responses against myelinating oligodendrocytes may contribute to neurodegenerative diseases with amyloidosis. © 2024. The Author(s).
- Cancer Research,
- Immunology and Microbiology
FLI1 promotes IFN-γ-induced kynurenine production to impair anti-tumor immunity.
In Nature Communications on 30 May 2024 by Chen, E., Wu, J., et al.
Nasopharyngeal carcinoma (NPC)-mediated immunosuppression within the tumor microenvironment (TME) frequently culminates in the failure of otherwise promising immunotherapies. In this study, we identify tumor-intrinsic FLI1 as a critical mediator in impairing T cell anti-tumor immunity. A mechanistic inquiry reveals that FLI1 orchestrates the expression of CBP and STAT1, facilitating chromatin accessibility and transcriptional activation of IDO1 in response to T cell-released IFN-γ. This regulatory cascade ultimately leads to augmented IDO1 expression, resulting in heightened synthesis of kynurenine (Kyn) in tumor cells. This, in turn, fosters CD8+ T cell exhaustion and regulatory T cell (Treg) differentiation. Intriguingly, we find that pharmacological inhibition of FLI1 effectively obstructs the CBP/STAT1-IDO1-Kyn axis, thereby invigorating both spontaneous and checkpoint therapy-induced immune responses, culminating in enhanced tumor eradication. In conclusion, our findings delineate FLI1-mediated Kyn metabolism as an immune evasion mechanism in NPC, furnishing valuable insights into potential therapeutic interventions. © 2024. The Author(s).
- Cancer Research,
- Immunology and Microbiology,
- Genetics
Genetic fusion of CCL11 to antigens enhances antigenicity in nucleic acid vaccines and eradicates tumor mass through optimizing T-cell response.
In Molecular Cancer on 8 March 2024 by Qi, H., Sun, Z., et al.
PubMed
Nucleic acid vaccines have shown promising potency and efficacy for cancer treatment with robust and specific T-cell responses. Improving the immunogenicity of delivered antigens helps to extend therapeutic efficacy and reduce dose-dependent toxicity. Here, we systematically evaluated chemokine-fused HPV16 E6/E7 antigen to improve the cellular and humoral immune responses induced by nucleotide vaccines in vivo. We found that fusion with different chemokines shifted the nature of the immune response against the antigens. Although a number of chemokines were able to amplify specific CD8 + T-cell or humoral response alone or simultaneously. CCL11 was identified as the most potent chemokine in improving immunogenicity, promoting specific CD8 + T-cell stemness and generating tumor rejection. Fusing CCL11 with E6/E7 antigen as a therapeutic DNA vaccine significantly improved treatment effectiveness and caused eradication of established large tumors in 92% tumor-bearing mice (n = 25). Fusion antigens with CCL11 expanded the TCR diversity of specific T cells and induced the infiltration of activated specific T cells, neutrophils, macrophages and dendritic cells (DCs) into the tumor, which created a comprehensive immune microenvironment lethal to tumor. Combination of the DNA vaccine with anti-CTLA4 treatment further enhanced the therapeutic effect. In addition, CCL11 could also be used for mRNA vaccine design. To summarize, CCL11 might be a potent T cell enhancer against cancer. © 2024. The Author(s).
- Mus musculus (House mouse)
The efficacy of chemotherapy is limited by intratumoral senescent cells expressing PD-L2.
In Nature Cancer on 1 March 2024 by Chaib, S., Lopez-Dominguez, J. A., et al.
Chemotherapy often generates intratumoral senescent cancer cells that strongly modify the tumor microenvironment, favoring immunosuppression and tumor growth. We discovered, through an unbiased proteomics screen, that the immune checkpoint inhibitor programmed cell death 1 ligand 2 (PD-L2) is highly upregulated upon induction of senescence in different types of cancer cells. PD-L2 is not required for cells to undergo senescence, but it is critical for senescent cells to evade the immune system and persist intratumorally. Indeed, after chemotherapy, PD-L2-deficient senescent cancer cells are rapidly eliminated and tumors do not produce the senescence-associated chemokines CXCL1 and CXCL2. Accordingly, PD-L2-deficient pancreatic tumors fail to recruit myeloid-derived suppressor cells and undergo regression driven by CD8 T cells after chemotherapy. Finally, antibody-mediated blockade of PD-L2 strongly synergizes with chemotherapy causing remission of mammary tumors in mice. The combination of chemotherapy with anti-PD-L2 provides a therapeutic strategy that exploits vulnerabilities arising from therapy-induced senescence. © 2024. The Author(s).
- Mus musculus (House mouse),
- Cancer Research
INHBA/Activin A promotes tumor growth and induces resistance to anti-PD-L1 therapy by suppressing IFN-γ signaling
Preprint on BioRxiv : the Preprint Server for Biology on 8 December 2023 by Li, F., Gu, L., et al.
PubMed
Inhibin beta A (INHBA) and its homodimer activin A have pleiotropic effects on modulation of immune responses and tumor progression, respectively, but it remains uncertain whether tumors may release activin A to regulate anti-tumor immunity. As evidenced by our RNA-Seq and in vitro results, the interferon-γ (IFN-γ) signaling pathway was significantly down-regulated by tumor intrinsic activin A. Tumor INHBA deficiency led to lower expression of PD-L1 induced by IFN-γ, resulting in poor responsiveness to anti-PD-L1 therapy. On the other hand, decreased secretion of IFN-γ-stimulated chemokines, including C-X-C motif chemokine 9 (CXCL9) and 10 (CXCL10), impaired the infiltration of effector T cells into the tumor microenvironment. Furthermore, the activin A-specific antibody garetosmab improved anti-tumor immunity and its combination with the anti-PD-L1 antibody atezolizumab showed a superior therapeutic effect to monotherapy. Our findings reveal that INHBA/activin A is involved in anti-tumor immunity by inhibiting the IFN-γ signaling pathway and considered to be a potential target to overcome anti-PD-L1 resistance in clinical cancer treatment.
- Cancer Research,
- Immunology and Microbiology
CTLA-4 blockade induces tumor pyroptosis via CD8+ T cells in head and neck squamous cell carcinoma.
In Molecular Therapy on 5 July 2023 by Wang, S., Wu, Z. Z., et al.
Immune checkpoint blockade (ICB) treatment has demonstrated excellent medical effects in oncology, and it is one of the most sought after immunotherapies for tumors. However, there are several issues with ICB therapy, including low response rates and a lack of effective efficacy predictors. Gasdermin-mediated pyroptosis is a typical inflammatory death mode. We discovered that increased expression of gasdermin protein was linked to a favorable tumor immune microenvironment and prognosis in head and neck squamous cell carcinoma (HNSCC). We used the mouse HNSCC cell lines 4MOSC1 (responsive to CTLA-4 blockade) and 4MOSC2 (resistant to CTLA-4 blockade) orthotopic models and demonstrated that CTLA-4 blockade treatment induced gasdermin-mediated pyroptosis of tumor cells, and gasdermin expression positively correlated to the effectiveness of CTLA-4 blockade treatment. We found that CTLA-4 blockade activated CD8+ T cells and increased the levels of interferon γ (IFN-γ) and tumor necrosis factor α (TNF-α) cytokines in the tumor microenvironment. These cytokines synergistically activated the STAT1/IRF1 axis to trigger tumor cell pyroptosis and the release of large amounts of inflammatory substances and chemokines. Collectively, our findings revealed that CTLA-4 blockade triggered tumor cells pyroptosis via the release of IFN-γ and TNF-α from activated CD8+ T cells, providing a new perspective of ICB. Copyright © 2023 The American Society of Gene and Cell Therapy. Published by Elsevier Inc. All rights reserved.
- Biochemistry and Molecular biology,
- Cancer Research,
- Cell Biology,
- Endocrinology and Physiology,
- Mus musculus (House mouse)
Transcriptional control of pancreatic cancer immunosuppression by metabolic enzyme CD73 in a tumor-autonomous and -autocrine manner.
In Nature Communications on 8 June 2023 by Tang, T., Huang, X., et al.
PubMed
Cancer cell metabolism contributes to the establishment of an immunosuppressive tumor microenvironment. Aberrant expression of CD73, a critical enzyme in ATP metabolism, on the cell surface results in the extracellular accumulation of adenosine, which exhibits direct inhibitory effects on tumor-infiltrating lymphocytes. However, little is known about the influence of CD73 on negative immune regulation-associated signaling molecules and transduction pathways inside tumor cells. This study aims to demonstrate the moonlighting functions of CD73 in immunosuppression in pancreatic cancer, an ideal model characterized by complex crosstalk among cancer metabolism, immune microenvironment, and immunotherapeutic resistance. The synergistic effect of CD73-specific drugs in combination with immune checkpoint blockade is observed in multiple pancreatic cancer models. Cytometry by time-of-flight analysis shows that CD73 inhibition reduces tumor-infiltrating Tregs in pancreatic cancer. Tumor cell-autonomous CD73 is found to facilitate Treg recruitment, in which CCL5 is identified as a significant downstream effector of CD73 using integrated proteomic and transcriptomic analyses. CD73 transcriptionally upregulates CCL5 through tumor cell-autocrine adenosine-Adora2a signaling-mediated activation of the p38-STAT1 axis, recruiting Tregs to pancreatic tumors and causing an immunosuppressive microenvironment. Together, this study highlights that CD73-adenosine metabolism transcriptionally controls pancreatic cancer immunosuppression in a tumor-autonomous and -autocrine manner. © 2023. The Author(s).
- Immunology and Microbiology
Vector Aided Microenvironment programming (VAMP): reprogramming the TME with MVA virus expressing IL-12 for effective antitumor activity.
In Journal for Immunotherapy of Cancer on 1 April 2023 by Seclì, L., Infante, L., et al.
PubMed
Tumor microenvironment (TME) represents a critical hurdle in cancer immunotherapy, given its ability to suppress antitumor immunity. Several efforts are made to overcome this hostile TME with the development of new therapeutic strategies modifying TME to boost antitumor immunity. Among these, cytokine-based approaches have been pursued for their known immunomodulatory effects on different cell populations within the TME. IL-12 is a potent pro-inflammatory cytokine that demonstrates striking immune activation and tumor control but causes severe adverse effects when systemically administered. Thus, local administration is considered a potential strategy to achieve high cytokine concentrations at the tumor site while sparing systemic adverse effects. Modified Vaccinia Ankara (MVA) vector is a potent inducer of pro-inflammatory response. Here, we cloned IL-12 into the genome of MVA for intratumoral immunotherapy, combining the immunomodulatory properties of both the vector and the cargo. The antitumor activity of MVA-IL-12 and its effect on TME reprogramming were investigated in preclinical tumor models. RNA sequencing (RNA-Seq) analysis was performed to assess changes in the TME in treated and distal tumors and the effect on the intratumoral T-cell receptor repertoire. Intratumoral injection of MVA-IL-12 resulted in strong antitumor activity with the complete remission of established tumors in multiple murine models, including those resistant to checkpoint inhibitors. The therapeutic activity of MVA-IL-12 was associated with very low levels of circulating cytokine. Effective TME reprogramming was demonstrated on treatment, with the reduction of immunosuppressive M2 macrophages while increasing pro-inflammatory M1, and recruitment of dendritic cells. TME switch from immunosuppressive into immunostimulatory environment allowed for CD8 T cells priming and expansion leading to tumor attack. Intratumoral administration of MVA-IL-12 turns immunologically 'cold' tumors 'hot' and overcomes resistance to programmed cell death protein-1 blockade. © Author(s) (or their employer(s)) 2023. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
- Cancer Research
Suppression of Tumor or Host Intrinsic CMTM6 Drives Antitumor Cytotoxicity in a PD-L1-Independent Manner.
In Cancer Immunology Research on 3 February 2023 by Long, Y., Chen, R., et al.
PubMed
CKLF-like MARVEL transmembrane domain-containing protein 6 (CMTM6) is known to be a regulator of membranal programmed death ligand 1 (PD-L1) stability and a factor associated with malignancy progression, but the effects and mechanisms of CMTM6 on tumor growth, as well as its potential as a target for therapy, are still largely unknown. Here, we show that CMTM6 expression increased with tumor progression in both patients and mice. Ablation of CMTM6 significantly reduced human and murine tumor growth in a manner dependent on T-cell immunity. Tumor CMTM6 suppression broke resistance to immune-checkpoint inhibitors and remodeled the tumor immune microenvironment, as specific antitumor cytotoxicity was enhanced and contributed primarily to tumor inhibition. Without the PD-1/PD-L1 axis, CMTM6 suppression still significantly dampened tumor growth dependent on cytotoxic cells. Furthermore, we identified that CMTM6 was widely expressed on immune cells. T-cell CMTM6 levels increased with sustained immune activation and intratumoral immune exhaustion and affected T cell-intrinsic PD-L1 levels. Host CMTM6 knockout significantly restrained tumor growth in a manner dependent on CD8+ T cells and not entirely dependent on PD-L1. Thus, we developed and evaluated the antitumor efficacy of CMTM6-targeting adeno-associated virus (AAV), which effectively mobilized antitumor immunity and could be combined with various antitumor drugs. Our findings reveal that both tumor and host CMTM6 are involved in antitumor immunity with or without the PD-1/PD-L1 axis and that gene therapy targeting CMTM6 is a promising strategy for cancer immunotherapy. ©2022 The Authors; Published by the American Association for Cancer Research.
- Mus musculus (House mouse)
The efficacy of chemotherapy is limited by intratumoural senescent cells that persist through the upregulation of PD-L2
Preprint on BioRxiv : the Preprint Server for Biology on 4 November 2022 by Chaib, S., López-Domínguez, J. A., et al.
PubMed
Anti-cancer therapies often result in a subset of surviving cancer cells that undergo therapy-induced senescence (TIS). Senescent cancer cells strongly modify the intratumoural microenvironment favoring immunosuppression and, thereby, tumour growth. An emerging strategy to optimise current therapies is to combine them with treatments that eliminate senescent cells. To this end, we undertook an unbiased proteomics approach to identify surface markers contributing to senescent cells immune evasion. Through this approach, we discovered that the immune checkpoint inhibitor PD-L2, but not PD-L1, is upregulated across multiple senescent human and murine cells. Importantly, blockade of PD-L2 strongly synergises with genotoxic chemotherapy, causing remission of solid tumours in mice. We show that PD-L2 inhibition prevents the persistence of chemotherapy-induced senescent cells, which exert cell-extrinsic immunomodulatory actions. In particular, upon chemotherapy, tumours deficient in PD-L2 fail to produce cytokines of the CXCL family, do not recruit myeloid-derived suppressor cells (MDSCs) and are eliminated in a CD8 T cell-dependent manner. We conclude that blockade of PD-L2 improves chemotherapy efficacy by reducing the intratumoural burden of senescent cells and their associated recruitment of immunosuppressive cells. These findings provide a novel strategy to exploit vulnerabilities arising in tumour cells as a result of therapy-induced damage and cellular senescence.
- Cancer Research,
- Immunology and Microbiology
The TLR7/8 agonist R848 optimizes host and tumor immunity to improve therapeutic efficacy in murine lung cancer.
In International Journal of Oncology on 1 July 2022 by Zhou, J., Xu, Y., et al.
PubMed
Treatment with the Toll‑like receptor 7 (TLR7) agonist, resiquimod (R848), is effective in various types of cancer, such as breast, pancreatic and colorectal cancer. The reported antitumor effect of R848 in lung cancer is considered to be achieved by targeting macrophages. In the present study, it was demonstrated that TLR7 expression on various immune cell types initially rises, then declines in the late stage of lung cancer. Intraperitoneal injection of R848 resulted in a reduction in tumor burden and prolonged survival in both subcutaneous and metastatic lung cancer models in C57BL/6 mice. Initial treatment with R848 at an early stage was found to be the optimal choice. Systemic injection of R848 promoted the activation of innate and adaptive immune responses. Systemic administration of R848 upregulated TLR7 expression in dendritic cells (DCs) and enhanced the activation of DCs and natural killer (NK) cells. Moreover, this treatment also resulted in increased production of T helper cell‑associated cytokines in serum, including IFN‑γ, TNF‑α and IL‑2. In addition, continuous treatment with R848 increased the proportion of DCs, NK and CD8+ T cells, and reduced that of Foxp3+ regulatory T cells in the tumor microenvironment. These findings supported the use of R848 treatment for lung cancer via TLR7 targeting and provided insight into the underlying therapeutic mechanism.
- In Vivo,
- Mus musculus (House mouse)
Ursodeoxycholic acid reduces antitumor immunosuppression by inducing CHIP-mediated TGF-β degradation.
In Nature Communications on 14 June 2022 by Shen, Y., Lu, C., et al.
PubMed
TGF-β is essential for inducing systemic tumor immunosuppression; thus, blocking TGF-β can greatly enhance antitumor immunity. However, there are still no effective TGF-β inhibitors in clinical use. Here, we show that the clinically approved compound ursodeoxycholic acid (UDCA), by degrading TGF-β, enhances antitumor immunity through restraining Treg cell differentiation and activation in tumor-bearing mice. Furthermore, UDCA synergizes with anti-PD-1 to enhance antitumor immunity and tumor-specific immune memory in tumor-bearing mice. UDCA phosphorylates TGF-β at T282 site via TGR5-cAMP-PKA axis, causing increased binding of TGF-β to carboxyl terminus of Hsc70-interacting protein (CHIP). Then, CHIP ubiquitinates TGF-β at the K315 site, initiating p62-dependent autophagic sorting and subsequent degradation of TGF-β. Notably, results of retrospective analysis shows that combination therapy with anti-PD-1 or anti-PD-L1 and UDCA has better efficacy in tumor patients than anti-PD-1 or anti-PD-L1 alone. Thus, our results show a mechanism for TGF-β regulation and implicate UDCA as a potential TGF-β inhibitor to enhance antitumor immunity. © 2022. The Author(s).
- Cancer Research,
- Immunology and Microbiology
NOTCH-Induced MDSC Recruitment after oHSV Virotherapy in CNS Cancer Models Modulates Antitumor Immunotherapy.
In Clinical Cancer Research on 1 April 2022 by Otani, Y., Yoo, J. Y., et al.
PubMed
Oncolytic herpes simplex virus-1 (oHSV) infection of brain tumors activates NOTCH, however the consequences of NOTCH on oHSV-induced immunotherapy is largely unknown. Here we evaluated the impact of NOTCH blockade on virus-induced immunotherapy. RNA sequencing (RNA-seq), TCGA data analysis, flow cytometry, Luminex- and ELISA-based assays, brain tumor animal models, and serum analysis of patients with recurrent glioblastoma (GBM) treated with oHSV was used to evaluate the effect of NOTCH signaling on virus-induced immunotherapy. TCGA data analysis of patients with grade IV glioma and oHSV treatment of experimental brain tumors in mice showed that NOTCH signaling significantly correlated with a higher myeloid cell infiltration. Immunofluorescence staining and RNA-seq uncovered a significant induction of Jag1 (NOTCH ligand) expression in infiltrating myeloid cells upon oHSV infection. Jag1-expressing macrophages further spread NOTCH activation in the tumor microenvironment (TME). NOTCH-activated macrophages increased the secretion of CCL2, which further amplified myeloid-derived suppressor cells. CCL2 and IL10 induction was also observed in serum of patients with recurrent GBM treated with oHSV (rQnestin34.5; NCT03152318). Pharmacologic blockade of NOTCH signaling rescued the oHSV-induced immunosuppressive TME and activated a CD8-dependent antitumor memory response, resulting in a therapeutic benefit. NOTCH-induced immunosuppressive myeloid cell recruitment limited antitumor immunity. Translationally, these findings support the use of NOTCH inhibition in conjunction with oHSV therapy. ©2022 American Association for Cancer Research.
- Cancer Research,
- Immunology and Microbiology,
- Mus musculus (House mouse)
Lacticaseibacillus paracasei sh2020 induced antitumor immunity and synergized with anti-programmed cell death 1 to reduce tumor burden in mice.
In Gut Microbes on 9 March 2022 by Zhang, S. L., Han, B., et al.
PubMed
The gut microbiota was emerging as critical regulatory elements in shaping the outcome of cancer immunotherapy. However, the underlying mechanisms by which the gut commensal species enhance antitumor immunity remain largely unexplored. Here, we show that the gut microbiota from healthy individuals conferred considerable sensitivity to anti-PD-1 in the colorectal cancer (CRC) tumor-bearing mice, whereas gut microbiota from CRC patients failed to do so. By 16S rRNA gene sequencing, we identified Lactobacillus that was significantly increased in the mice with good response to anti-PD-1, and significantly correlated with anti-tumor immunity. After a series of screening, we isolated a novel Lacticaseibacillus strain, named L. paracasei sh2020. L. paracasei sh2020 showed the most notable anti-tumor immunity in the mice with gut dysbiosis. Mechanistically, the antitumor immune response elicited by L. paracasei sh2020 was dependent on CD8+ T cell. In vitro and in vivo studies revealed that L. paracasei sh2020 stimulation triggered the upregulated expression of CXCL10 in the tumors and subsequently enhanced CD8+ T cell recruitment. Meanwhile, the modulation of gut microbiota caused by L. paracasei sh2020 enhanced its antitumor effect and gut barrier function. Overall, our study offered novel insights into the mechanism by which gut microbiota shaped the outcome of cancer immunotherapy and, more importantly, the novel strain L. paracasei sh2020 might serve as an easy and effective way to promote anti-PD-1 effect in clinical practice.