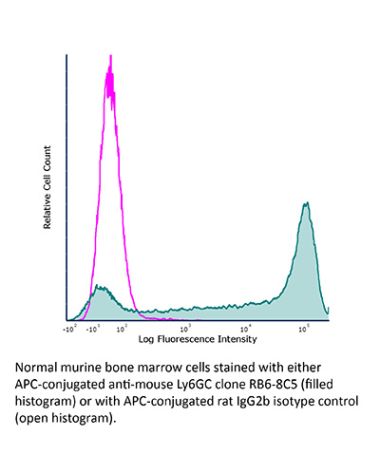
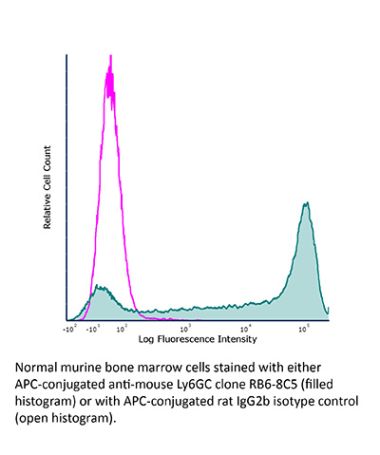
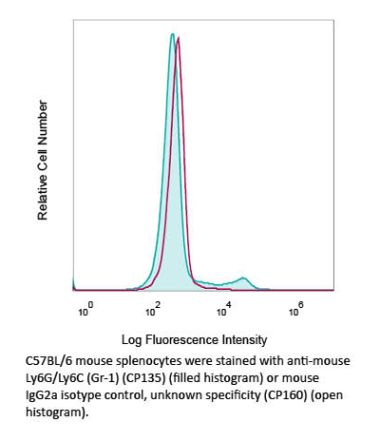
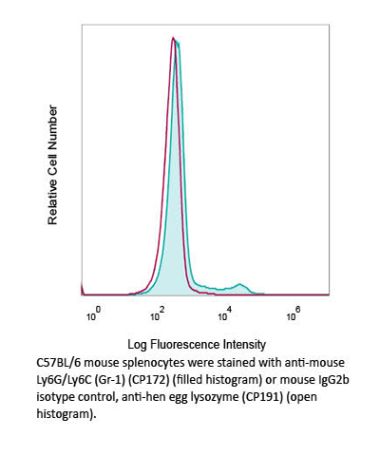
InVivoMAb anti-mouse Ly6G/Ly6C
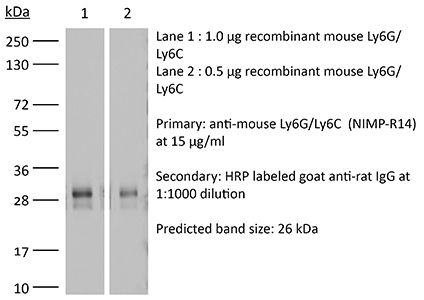
Product Details
The NIMP-R14 monoclonal antibody is specific for murine neutrophils. The antibody is reported to react strongly with mouse Ly6G and Ly6C previously referred to as GR-1. Ly6G is a 21-25 kDa member of the Ly-6 superfamily of GPI-anchored cell surface proteins with roles in cell signaling and cell adhesion. Ly6G is expressed differentially during development by cells in the myeloid lineage including monocytes, macrophages, granulocytes, and neutrophils. Monocytes typically express Ly6G transiently during development while mature granulocytes and peripheral neutrophils retain expression making Ly6G a good cell surface marker for these populations. Ly6C is a monocyte/macrophage and endothelial cell differentiation antigen regulated by interferon gamma and is thought to play a role in the development and maturation of lymphocytes. The NIMP-R14 antibody has been shown to be useful for depletion of neutrophils in mice.Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Purified BALB/c mouse neutrophils |
| Reported Applications |
in vivo neutrophil depletion Immunohistochemistry (paraffin) Immunohistochemistry (frozen) Immunofluorescence Flow cytometry |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
<2EU/mg (<0.002EU/μg) Determined by LAL gel clotting assay |
| Purity |
>95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein A |
| RRID | AB_2819047 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Recommended Products
-

Recommended Isotype Control(s)
InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin
-

Recommended Dilution Buffer
InVivoPure pH 7.0 Dilution Buffer
in vivo neutrophil depletion
Castell, S. D., et al. (2019). "Neutrophils Which Migrate to Lymph Nodes Modulate CD4(+) T Cell Response by a PD-L1 Dependent Mechanism" Front Immunol 10: 105. PubMed
It is well known that neutrophils are rapidly recruited to a site of injury or infection and perform a critical role in pathogen clearance and inflammation. However, they are also able to interact with and regulate innate and adaptive immune cells and some stimuli induce the migration of neutrophils to lymph nodes (LNs). Previously, we demonstrated that the immune complex (IC) generated by injecting OVA into the footpad of OVA/CFA immunized mice induced the migration of OVA(+) neutrophils to draining LNs (dLNs). Here we investigate the effects of these neutrophils which reach dLNs on CD4(+) T cell response. Our findings here strongly support a dual role for neutrophils in dLNs regarding CD4(+) T cell response modulation. On the one hand, the CD4(+) T cell population expands after the influx of OVA(+) neutrophils to dLNs. These CD4(+) T cells enlarge their proliferative response, activation markers and IL-17 and IFN-gamma cytokine production. On the other hand, these neutrophils also restrict CD4(+) T cell expansion. The neutrophils in the dLNs upregulate PD-L1 molecules and are capable of suppressing CD4(+) T cell proliferation. These results indicate that neutrophils migration to dLNs have an important role in the homeostasis of adaptive immunity. This report describes for the first time that the influx of neutrophils to dLNs dependent on IC presence improves CD4(+) T cell response, at the same time controlling CD4(+) T cell proliferation through a PD-L1 dependent mechanism.
in vivo neutrophil depletion
Stackowicz, J., et al. (2018). "Evidence that neutrophils do not promote Echis carinatus venom-induced tissue destruction" Nat Commun 9(1): 2304. PubMed
in vivo neutrophil depletion
Karmakar, M., et al. (2016). "Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1beta secretion in response to ATP" Nat Commun 7: 10555. PubMed
Although extracellular ATP is abundant at sites of inflammation, its role in activating inflammasome signalling in neutrophils is not well characterized. In the current study, we demonstrate that human and murine neutrophils express functional cell-surface P2X7R, which leads to ATP-induced loss of intracellular K(+), NLRP3 inflammasome activation and IL-1beta secretion. ATP-induced P2X7R activation caused a sustained increase in intracellular [Ca(2+)], which is indicative of P2X7R channel opening. Although there are multiple polymorphic variants of P2X7R, we found that neutrophils from multiple donors express P2X7R, but with differential efficacies in ATP-induced increase in cytosolic [Ca(2+)]. Neutrophils were also the predominant P2X7R-expressing cells during Streptococcus pneumoniae corneal infection, and P2X7R was required for bacterial clearance. Given the ubiquitous presence of neutrophils and extracellular ATP in multiple inflammatory conditions, ATP-induced P2X7R activation and IL-1beta secretion by neutrophils likely has a significant, wide ranging clinical impact.
in vivo neutrophil depletion, Flow Cytometry
Karmakar, M., et al. (2012). "Cutting edge: IL-1beta processing during Pseudomonas aeruginosa infection is mediated by neutrophil serine proteases and is independent of NLRC4 and caspase-1" J Immunol 189(9): 4231-4235. PubMed
To examine the role of caspase-1 and the NLRC4 inflammasome during bacterial infection, C57BL/6, IL-1beta(-/-), caspase-1(-/-), and NLRC4(-/-) mouse corneas were infected with ExoS/T- or ExoU-expressing Pseudomonas aeruginosa. We found that IL-1beta was essential for neutrophil recruitment and bacterial clearance and was produced by myeloid cells rather than resident cells. In addition, neutrophils were found to be the primary source of mature IL-1beta during infection, and there was no significant difference in IL-1beta processing between C57BL/6 and caspase-1(-/-) or NLRC4(-/-) infected corneas. IL-1beta cleavage by human and mouse neutrophils was blocked by serine protease inhibitors and was impaired in infected neutrophil elastase (NE)(-/-) corneas. NE(-/-) mice also had an impaired ability to clear the infection. Together, these results demonstrate that during P. aeruginosa infection, neutrophils are the primary source of mature IL-1beta and that IL-1beta processing is dependent on serine proteases and not NLRC4 or caspase-1.
Immunofluorescence
Chung, E. S., et al. (2009). "Contribution of macrophages to angiogenesis induced by vascular endothelial growth factor receptor-3-specific ligands" Am J Pathol 175(5): 1984-1992. PubMed
Vascular endothelial growth factor receptor (VEGFR)-2 is a major stimulator of hemangiogenesis (HA), whereas VEGFR-3 stimulates lymphangiogenesis (LA). Contrary to this understanding, we demonstrate that implantation of pellets containing VEGFR-3-specific ligands (VEGF-C156S and recombinant murine VEGF-D) into the corneal stroma induce not only LA but also robust HA characterized by blood vessels that are positive for VEGFR-3 expression. The implantation of pellets containing VEGFR-3-specific ligands also leads to the recruitment of VEGF-A-secreting macrophages. Depletion of these infiltrating macrophages using clodronate-liposome administration shows a significant reduction in HA as well as LA. Blockade of either VEGFR-2 or VEGFR-3 signaling reduces both HA and LA; however, the percent reduction of HA is greater in the VEGFR-2 blockade group. In addition, in the VEGFR-3 blockade group, the percent reduction of HA is significantly greater with VEGFR-3-specific ligands than that by VEGF-A or VEGF-C. Collectively, our data suggest that VEGFR-3-specific signaling can induce new blood vessels, to which macrophages contribute a major role, and signify its potential as an additional therapeutic target to the existing VEGF-A/VEGFR-2 signaling-based antiangiogenesis strategies.
Immunohistochemistry (paraffin)
Abdollahi-Roodsaz, S., et al. (2009). "Local interleukin-1-driven joint pathology is dependent on toll-like receptor 4 activation" Am J Pathol 175(5): 2004-2013. PubMed
Toll-like receptors (TLRs) may contribute to the pathogenesis of chronic inflammatory destructive diseases through the recognition of endogenous ligands produced on either inflammation or degeneration of the extracellular matrix. The presence of endogenous TLR agonists has been reported in rheumatoid joints. In the present study, we investigated the significance of TLR2 and TLR4 activation by locally- produced endogenous ligands in the severity of joint inflammation and destruction. Local joint pathology independent of systemic immune activation was induced by overexpression of interleukin (IL)-1 and TNF in naive joints using adenoviral gene transfer. Here, we report that at certain doses, IL-1-induced local joint inflammation, cartilage proteoglycan depletion, and bone erosion are dependent on TLR4 activation, whereas TLR2 activation is not significantly involved. In comparison, tumor necrosis factor alpha-driven joint pathology seemed to be less dependent on TLR2 and TLR4. The severity of IL-1-induced bone erosion and irreversible cartilage destruction was markedly reduced in TLR4(-/-) mice, even though the degree of inflammation was similar, suggesting uncoupled processes. Furthermore, the expression of cathepsin K, a marker for osteoclast activity, induced by IL-1beta was dependent on TLR4. Overexpression of IL-1beta in the joint as well as ex vivo IL-1 stimulation of patellae provoked the release of endogenous TLR4 agonists capable of inducing TLR4-mediated cytokine production. These data emphasize the potential relevance of TLR4 activation in rheumatoid arthritis, particularly with respect to IL-1-mediated joint pathology.
Immunohistochemistry (frozen)
Carlson, E. C., et al. (2007). "Keratocan and lumican regulate neutrophil infiltration and corneal clarity in lipopolysaccharide-induced keratitis by direct interaction with CXCL1" J Biol Chem 282(49): 35502-35509. PubMed
Keratocan and lumican are keratan-sulfate proteoglycans (KSPG), which have a critical role in maintaining corneal clarity. To determine whether these KSPGs have a role in corneal inflammation, we examined Kera(-/-) and Lum(-/-) mice in a model of lipopolysaccharide (LPS)-induced keratitis in which wild-type mice develop increased corneal thickness and haze due to neutrophil infiltration to the corneal stroma. Corneal thickness increases caused by LPS mice were significantly lower in Kera(-/-) and Lum(-/-) than wild-type mice. Further, LPS-injected Lum(-/-) mice had elevated corneal haze levels compared with that of Kera(-/-) and wild-type. At 24 h post-injection, total enhanced green fluorescent protein-positive bone marrow-derived inflammatory cells in chimeric mice was significantly lower in Kera(-/-) mice and Lum(-/-) mice compared with wild-type mice. Neutrophil infiltration was inhibited in Kera(-/-) and Lum(-/-) mice at 6 and 24 h post-stimulation, with Lum(-/-) corneas having the most profound defect in neutrophil migration. Reconstitution of keratocan and lumican expression in corneas of Kera(-/-) and Lum(-/-) mice using adeno-keratocan and adeno-lumican viral vectors, respectively, resulted in normal neutrophil infiltration in response to LPS. Immunoprecipitation/Western blot analysis showed that lumican and keratocan core proteins bind the CXC chemokine KC during a corneal inflammatory response, indicating that corneal KSPGs mediate neutrophil recruitment to the cornea by regulating chemokine gradient formation. Together, these data support a significant role for lumican and keratocan in a corneal inflammatory response with respect to edema, corneal clarity, and cellular infiltration.
- Mus musculus (House mouse),
- Immunology and Microbiology
West Nile virus triggers intestinal dysmotility via T cell-mediated enteric nervous system injury.
In The Journal of Clinical Investigation on 29 August 2024 by Janova, H., Zhao, F. R., et al.
Intestinal dysmotility syndromes have been epidemiologically associated with several antecedent bacterial and viral infections. To model this phenotype, we previously infected mice with the neurotropic flavivirus West Nile virus (WNV) and demonstrated intestinal transit defects. Here, we found that within 1 week of WNV infection, enteric neurons and glia became damaged, resulting in sustained reductions of neuronal cells and their networks of connecting fibers. Using cell-depleting antibodies, adoptive transfer experiments, and mice lacking specific immune cells or immune functions, we show that infiltrating WNV-specific CD4+ and CD8+ T cells damaged the enteric nervous system (ENS) and glia, which led to intestinal dysmotility; these T cells used multiple and redundant effector molecules including perforin and Fas ligand. In comparison, WNV-triggered ENS injury and intestinal dysmotility appeared to not require infiltrating monocytes, and damage may have been limited by resident muscularis macrophages. Overall, our experiments support a model in which antigen-specific T cell subsets and their effector molecules responding to WNV infection direct immune pathology against enteric neurons and supporting glia that results in intestinal dysmotility.
- Mus musculus (House mouse),
- Cancer Research,
- Immunology and Microbiology
Heterodimerization of T cell engaging bispecific antibodies to enhance specificity against pancreatic ductal adenocarcinoma.
In Journal of Hematology & Oncology on 23 April 2024 by Long, A. W., Xu, H., et al.
EGFR and/or HER2 expression in pancreatic cancers is correlated with poor prognoses. We generated homodimeric (EGFRxEGFR or HER2xHER2) and heterodimeric (EGFRxHER2) T cell-engaging bispecific antibodies (T-BsAbs) to direct polyclonal T cells to these antigens on pancreatic tumors. EGFR and HER2 T-BsAbs were constructed using the 2 + 2 IgG-[L]-scFv T-BsAbs format bearing two anti-CD3 scFvs attached to the light chains of an IgG to engage T cells while retaining bivalent binding to tumor antigens with both Fab arms. A Fab arm exchange strategy was used to generate EGFRxHER2 heterodimeric T-BsAb carrying one Fab specific for EGFR and one for HER2. EGFR and HER2 T-BsAbs were also heterodimerized with a CD33 control T-BsAb to generate 'tumor-monovalent' EGFRxCD33 and HER2xCD33 T-BsAbs. T-BsAb avidity for tumor cells was studied by flow cytometry, cytotoxicity by T-cell mediated 51Chromium release, and in vivo efficacy against cell line-derived xenografts (CDX) or patient-derived xenografts (PDX). Tumor infiltration by T cells transduced with luciferase reporter was quantified by bioluminescence. The EGFRxEGFR, HER2xHER2, and EGFRxHER2 T-BsAbs demonstrated high avidity and T cell-mediated cytotoxicity against human pancreatic ductal adenocarcinoma (PDAC) cell lines in vitro with EC50s in the picomolar range (0.17pM to 18pM). They were highly efficient in driving human polyclonal T cells into subcutaneous PDAC xenografts and mediated potent T cell-mediated anti-tumor effects. Both EGFRxCD33 and HER2xCD33 tumor-monovalent T-BsAbs displayed substantially reduced avidity by SPR when compared to homodimeric EGFRxEGFR or HER2xHER2 T-BsAbs (∼150-fold and ∼6000-fold respectively), tumor binding by FACS (8.0-fold and 63.6-fold), and T-cell mediated cytotoxicity (7.7-fold and 47.2-fold), while showing no efficacy against CDX or PDX. However, if either EGFR or HER2 was removed from SW1990 by CRISPR-mediated knockout, the in vivo efficacy of heterodimeric EGFRxHER2 T-BsAb was lost. EGFR and HER2 were useful targets for driving T cell infiltration and tumor ablation. Two arm Fab binding to either one or both targets was critical for robust anti-tumor effect in vivo. By engaging both targets, EGFRxHER2 heterodimeric T-BsAb exhibited potent anti-tumor effects if CDX or PDX were EGFR+HER2+ double-positive with the potential to spare single-positive normal tissue. © 2024. The Author(s).
- Mus musculus (House mouse),
- Immunology and Microbiology,
- Cancer Research
A CSF-1R-blocking antibody/IL-10 fusion protein increases anti-tumor immunity by effectuating tumor-resident CD8+ T cells.
In Cell Reports Medicine on 15 August 2023 by Chang, Y. W., Hsiao, H. W., et al.
PubMed
Strategies to increase intratumoral concentrations of an anticancer agent are desirable to optimize its therapeutic potential when said agent is efficacious primarily within a tumor but also have significant systemic side effects. Here, we generate a bifunctional protein by fusing interleukin-10 (IL-10) to a colony-stimulating factor-1 receptor (CSF-1R)-blocking antibody. The fusion protein demonstrates significant antitumor activity in multiple cancer models, especially head and neck cancer. Moreover, this bifunctional protein not only leads to the anticipated reduction in tumor-associated macrophages but also triggers proliferation, activation, and metabolic reprogramming of CD8+ T cells. Furthermore, it extends the clonotype diversity of tumor-infiltrated T cells and shifts the tumor microenvironment (TME) to an immune-active state. This study suggests an efficient strategy for designing immunotherapeutic agents by fusing a potent immunostimulatory molecule to an antibody targeting TME-enriched factors. Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
- Cancer Research
Immunosuppressive tumor microenvironment in uterine serous carcinoma via CCL7 signal with myeloid-derived suppressor cells.
In Carcinogenesis on 30 August 2022 by Mise, Y., Hamanishi, J., et al.
PubMed
Serous carcinoma of the uterus (USC) is a pathological subtype of high-grade endometrial cancers, with no effective treatment for advanced cases. Since such refractory tumors frequently harbor antitumor immune tolerance, many immunotherapies have been investigated for various malignant tumors using immuno-competent animal models mimicking their local immunities. In this study, we established an orthotopic mouse model of high-grade endometrial cancer and evaluated the local tumor immunity to explore the efficacy of immunotherapies against USC. A multivariate analysis of 62 human USC cases revealed that the tumor-infiltrating cell status, few CD8+ cells and abundant myeloid-derived suppressor cells (MDSCs), was an independent prognostic factor (P < 0.005). A murine endometrial cancer cell (mECC) was obtained from C57BL/6 mice via endometrium-specific deletion of Pten and Tp53, and another high-grade cell (HPmECC) was established by further overexpressing Myc in mECCs. HPmECCs exhibited higher capacities of migration and anchorage-independent proliferation than mECCs (P < 0.01, P < 0.0001), and when both types of cells were inoculated into the uterus of C57BL/6 mice, the prognosis of mice bearing HPmECC-derived tumors was significantly poorer (P < 0.001). Histopathological analysis of HPmECC orthotopic tumors showed serous carcinoma-like features with prominent tumor infiltration of MDSCs (P < 0.05), and anti-Gr-1 antibody treatment significantly prolonged the prognosis of HPmECC-derived tumor-bearing mice (P < 0.05). High CCL7 expression was observed in human USC and HPmECC, and MDSCs migration was promoted in a CCL7 concentration-dependent manner. These results indicate that antitumor immunity is suppressed in USC due to increased number of tumor-infiltrating MDSCs via CCL signal. © The Author(s) 2022. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
- In Vivo,
- Homo sapiens (Human),
- Cancer Research,
- Immunology and Microbiology
Modulating tumor infiltrating myeloid cells to enhance bispecific antibody-driven T cell infiltration and anti-tumor response.
In Journal of Hematology & Oncology on 8 September 2021 by Park, J. A., Wang, L., et al.
PubMed
Tumor microenvironment (TME) is a dynamic cellular milieu to promote tumor angiogenesis, growth, proliferation, and metastasis, while derailing the host anti-tumor response. TME impedes bispecific antibody (BsAb) or chimeric antigen receptor (CAR)-driven T cells infiltration, survival, and cytotoxic efficacy. Modulating tumor infiltrating myeloid cells (TIMs) could potentially improve the efficacy of BsAb. We evaluated the effects of TIM modulation on BsAb-driven T cell infiltration into tumors, their persistence, and in vivo anti-tumor response. Anti-GD2 BsAb and anti-HER2 BsAb built on IgG-[L]-scFv platform were tested against human cancer xenografts in BALB-Rag2-/-IL-2R-γc-KO (BRG) mice. Depleting antibodies specific for polymorphonuclear myeloid-derived suppressor cell (PMN-MDSC), monocytic MDSC (M-MDSC), and tumor associated macrophage (TAM) were used to study the role of each TIM component. Dexamethasone, an established anti-inflammatory agent, was tested for its effect on TIMs. BsAb-driven T cells recruited myeloid cells into human tumor xenografts. Each TIM targeting therapy depleted cells of interest in blood and in tumors. Depletion of PMN-MDSCs, M-MDSCs, and particularly TAMs was associated with enhanced T cell infiltration into tumors, significantly improving tumor control and survival in multiple cancer xenograft models. Dexamethasone premedication depleted monocytes in circulation and TAMs in tumors, enhanced BsAb-driven T cell infiltration, and anti-tumor response with survival benefit. Reducing TIMs markedly enhanced anti-tumor effects of BsAb-based T cell immunotherapy by improving intratumoral T cell infiltration and persistence. TAM depletion was more effective than PMN- or M-MDSCs depletion at boosting the anti-tumor response of T cell engaging BsAb. © 2021. The Author(s).
- Immunology and Microbiology
Immune Responses after Vascular Photodynamic Therapy with Redaporfin.
In Journal of Clinical Medicine on 31 December 2019 by Lobo, A. C. S., Gomes-da-Silva, L. C., et al.
PubMed
Photodynamic therapy (PDT) relies on the administration of a photosensitizer (PS) that is activated, after a certain drug-to-light interval (DLI), by the irradiation of the target tumour with light of a specific wavelength absorbed by the PS. Typically, low light doses are insufficient to eradicate solid tumours and high fluence rates have been described as poorly immunogenic. However, previous work with mice bearing CT26 tumours demonstrated that vascular PDT with redaporfin, using a low light dose delivered at a high fluence rate, not only destroys the primary tumour but also reduces the formation of metastasis, thus suggesting anti-tumour immunity. This work characterizes immune responses triggered by redaporfin-PDT in mice bearing CT26 tumours. Our results demonstrate that vascular-PDT leads to a strong neutrophilia (2-24 h), systemic increase of IL-6 (24 h), increased percentage of CD4+ and CD8+ T cells producing IFN-γ or CD69+ (2-24 h) and increased CD4+/CD8+ T cell ratio (2-24 h). At the tumour bed, T cell tumour infiltration disappeared after PDT but reappeared with a much higher incidence one day later. In addition, it is shown that the therapeutic effect of redaporfin-PDT is highly dependent on neutrophils and CD8+ T cells but not on CD4+ T cells.